Maladie de Joubert

La maladie de Joubert est une maladie rare d’origine génétique, et responsable d’une malformation au niveau du cervelet et du tronc cérébral. L’affection se manifeste dès la naissance par divers symptômes, associés ou non, comme une hypotonie, des difficultés respiratoires, des troubles oculaires et de l’équilibre. Le diagnostic est clinique, et confirmé par IRM. Il n’existe, actuellement, aucun traitement pour cette maladie. La prise en charge est symptomatique.
Définition de la maladie
Qu’est-ce que la maladie de Joubert ?
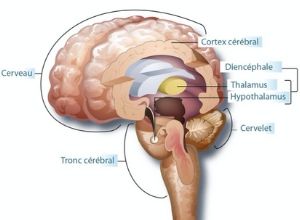
La maladie de Joubert est un syndrome génétique affectant le cervelet et le tronc cérébral qui sont deux organes localisés à la base du cerveau. Cette affection a été décrite pour la première fois en 1969 par les Docteurs Joubert et Eisenring.
Les premières manifestations de la maladie sont généralement détectées dès le plus jeune âge. Chez le nourrisson, la maladie se traduit par une hypotonie, des troubles respiratoires et des mouvements oculaires anormaux. L’enfant souffre, quant à lui, de divers troubles : de l’équilibre, de la coordination des mouvements, oculaires et d’apprentissage.
Les enfants atteints de la maladie de Joubert ont une malformation du système nerveux central qui implique une absence ou une diminution du volume de la partie médiane du cervelet. Or, le rôle du cervelet est de coordonner les mouvements. Son atteinte est donc à l’origine de l’hypotonie, des troubles de l’équilibre et des mouvements anormaux oculaires. L’atteinte du tronc cérébral, localisé juste en dessous du cervelet et qui héberge les centres nerveux contrôlant la respiration, est responsable des symptômes respiratoires.
Cette pathologie est rare. En effet, le nombre d’individus atteints de la maladie de Joubert est estimé à 1 sur 100 000. Elle touche aussi bien filles que garçons, peu importe leur origine géographique. Plusieurs anomalies (ou mutations) génétiques peuvent être à l’origine du trouble :
- Le gène NPHP1 localisé sur le chromosome 2 dans 1 à 2% des cas ;
- Le gène AHI1 que le chromosome 6 est concerné dans environ 11% des cas ;
- Le gène CEP290 situé sur le chromosome 12, dont la fréquence n’est pas encore connue.
D’autres gènes sont très certainement impliqués, mais les recherches sont encore en cours les concernant.
La maladie est dite « autosomique récessive ». Pour rappel, un chromosome est un élément microscopique constitué de molécules d’ADN. Il contient donc les gènes, supports de l’information génétique qui sont transmissibles à la descendance. Les chromosomes vont par paire. Tout individu sain dispose de 23 paires de chromosomes. La paire 23 contient les chromosomes sexuels qui déterminent le sexe de l’individu qui les porte : XX chez une femme, et XY chez un homme. Enfin, les chromosomes d’une même paire sont identiques, autrement dit, ils portent les mêmes gènes. Ainsi, il n’existe pas une version d’un même gène, mais deux, on parle d’allèle.
Le terme autosomique signifie que le gène impliqué n’est pas porté par un chromosome sexuel, mais par l’une des 22 autres paires de chromosomes. Ainsi, les deux sexes sont concernés de la même manière par la maladie.
On parle de transmission récessive lorsque l’anomalie génétique responsable de la maladie affecte les deux copies du gène impliqué. Autrement dit, un individu peut être porteur de l’anomalie (sur l’une des deux copies du gène) sans que celle-ci ne s’exprime. La maladie se manifeste uniquement lorsque les deux copies sont anormales. Ainsi, un individu porteur risque de transmettre la maladie à sa descendance si son partenaire est également porteur de l’anomalie.
Symptômes, diagnostic et traitement
Quels symptômes ?
- À la naissance
 La maladie de Joubert est généralement diagnostiquée dès la naissance. Elle se traduit, en effet, par divers symptômes caractéristiques comme une hypotonie, autrement dit, le nourrisson est mou, associée à un retard de développement moteur.
La maladie de Joubert est généralement diagnostiquée dès la naissance. Elle se traduit, en effet, par divers symptômes caractéristiques comme une hypotonie, autrement dit, le nourrisson est mou, associée à un retard de développement moteur.
À noter ! Les symptômes peuvent varier d’un individu à un autre. Il n’existe pas réellement de forme typique, mais plutôt des symptômes plus ou moins présents selon les cas. Aucun enfant n’a tous les symptômes décrits à la fois.
Le petit patient peut aussi présenter des troubles de la respiration : respiration irrégulière alternant entre apnées et accélération du rythme respiratoire. Ces troubles respiratoires peuvent nécessiter une prise en charge spécifique chez le nouveau-né. Cependant, avec l’âge, ils ont tendance à diminuer et disparaître.
Enfin, des mouvements oculaires anormaux peuvent être présents. On parle de nystagmus (mouvements oculaires saccadés).
- Dans l’enfance
Dans l’enfance, l’hypotonie associée aux troubles de l’équilibre, retarde les acquisitions motrices telles que la marche ou la station assise.
Chez certains petits patients, il est aussi décrit des mouvements anormaux de la langue.
Les troubles oculaires sont souvent présents et engendrent des difficultés à suivre du regard pour le patient. Il est fréquent d’avoir l’impression que l’enfant ne regarde pas bien en face, ou qu’il fuit le regard. Par ailleurs, les troubles oculaires peuvent être compensés par une inclinaison ou des secousses de la tête. Les symptômes oculaires s’améliorent avec en grandissant.
Il peut exister un déficit intellectuel plus ou moins important. Des troubles de l’élocution peuvent gêner la parole. Certains enfants sont parfois considérés comme autistes à tort. Quelques patients ont des crises convulsives.
Plus rarement, le petit patient peut souffrir :
- D’autres troubles oculaires comme une atteinte de la rétine (pouvant engendrer une cécité précoce), un strabisme, un ptôsis (chute des paupières), une mauvaise vision, etc. ;
- D’atteintes rénales ;
- De manifestations hépatiques (le foie est gros et fonctionne mal) ;
- De doigts surnuméraires. On parle de polydactylie ;
- De dysfonctionnement hormonal ;
- D’anomalies osseuses ;
- Etc.
L’évolution de la maladie varie beaucoup d’un patient à un autre. Les symptômes les plus gênants sont les troubles de l’équilibre, les symptômes oculaires et les difficultés d’apprentissage. Leur intensité est différente selon l’enfant. D’autres atteintes, par exemple l’atteinte rénale ou rétinienne, peuvent aboutir à des handicaps spécifiques.
Quel diagnostic ?
A la naissance, le diagnostic de la maladie de Joubert est clinique devant la présence des symptômes caractéristiques : hypotonie, troubles de l’équilibre, retard moteur, mouvements oculaires anormaux, troubles respiratoires.
Une IRM est nécessaire pour confirmer le diagnostic. Cet examen permet de visualiser les malformations du cervelet et du tronc cérébral.
À noter ! Malgré l’origine génétique de la maladie, une analyse génétique n’est pas indispensable au diagnostic.
Quel traitement ?
 Il n’existe aucun traitement permettant de guérir la maladie de Joubert. Une prise en charge symptomatique peut néanmoins soulager les patients et améliorer leur qualité de vie.
Il n’existe aucun traitement permettant de guérir la maladie de Joubert. Une prise en charge symptomatique peut néanmoins soulager les patients et améliorer leur qualité de vie.
L’hypotonie et le retard des acquisitions peuvent être améliorés par différentes méthodes de rééducation : la kinésithérapie, la psychomotricité, l’ergothérapie, l’orthophonie.
Les troubles oculaires sont pris en charge par un ophtalmologue et un orthoptiste : prescription de verres correcteurs, rééducation orthoptique, chirurgie (en cas de ptôsis ou de strabisme), etc.
Dans les rares cas où les manifestations respiratoires sont préoccupantes, il est possible d’équiper le patient d’un monitoring, un appareil permettant d’alerter l’entourage en cas d’apnée. Une prescription d’oxygène peut être associée. Cependant, en grandissant les symptômes respiratoires disparaissent dans la majorité des cas, et ne nécessitent donc plus aucune prise en charge.
En cas d’épilepsie, des antiépileptiques peuvent être prescrits, ou un traitement psychotrope en cas de trouble du comportement.
Les doigts surnuméraires peuvent être retirés chirurgicalement.
Enfin, un dépistage rénal et hépatique est recommandé.
Charline D., Docteur en pharmacie
Cet article vous a-t-il été utile ?