Syndrome de Marfan
Le syndrome de Marfan est une maladie génétique rare résultant d’une altération d’un des composants essentiels du tissu conjonctif : la fibrilline 1.
Ce syndrome se caractérise principalement par des atteintes des systèmes squelettique, oculaire et cardiovasculaire, mais d’autres organes, tels que la peau, les poumons et le système nerveux central peuvent aussi être touchés. La prise en charge de ces malades requiert par conséquent un suivi régulier et multidisciplinaire.
On compte une personne sur 5 000 atteinte de ce syndrome héréditaire.
Syndrome de Marfan : des causes génétiques
Le syndrome de Marfan (MFS) est une maladie génétique rare due à une production insuffisante d’une protéine, la fibrilline 1, protéine essentielle du tissu conjonctif. L’altération de cette protéine fait suite à une ou plusieurs mutations produites sur le gène codant pour cette protéine : le gène principal identifié est porté sur le chromosome 15, appelé FBN1 (plus de 600 mutations différentes affectant 66 à 91% des cas du syndrome de Marfan) mais dans 15% des cas, un autre gène serait aussi responsable tel que TGFBR2, localisé sur le chromosome 3. D’autres gènes pourraient aussi être concernés mais leur rôle dans la maladie n’a pas encore été précisément identifié.
A savoir ! Le tissu conjonctif est un tissu assurant la cohésion et le soutien des éléments d’un organe ou des organes entre eux. Les muscles, os, cartilages, ligaments et tendons sont essentiellement constitués de tissu conjonctif. Le tissu conjonctif est également présent dans la peau et les organes internes. Il s’agit d’un tissu très résistant capable de supporter le poids et les tensions.
Prévalence
Il s’agit d’une maladie héréditaire, à transmission autosomique dominante (transmission de génération en génération sans prédominance de sexe) touchant près d’une naissance sur 10 000 et dont la prévalence dans la population est de l’ordre de 1 sur 3 000 à 5 000 individus. Elle touche aussi bien les enfants que les adultes, homme et femmes.
Symptômes du syndrome de Marfan
Comme le tissu conjonctif est un tissu très largement distribué à travers le corps humain, le syndrome de Marfan peut toucher plusieurs organes différents et être à l’origine d’un grand nombre d’anomalies.
Chaque patient atteint du syndrome présente des symptômes cliniques qui lui sont personnels : un individu atteint peut présenter un tableau clinique différent d’un autre membre de la même famille. Ces symptômes cliniques apparaissent progressivement si bien que, chez l’enfant, il est souvent difficile de faire le diagnostic avant l’âge de 5 ans.
Par ailleurs, la fibrilline 1 est une protéine participant à la consolidation de différents tissus et organes. Etant défectueuse, les fonctions de soutien et de résistance du tissu conjonctif ne sont désormais plus assurées. Ceci entraîne des symptômes différents selon l’organe atteint.
Atteinte cardiovasculaire (cœur et vaisseaux sanguins)
L’atteinte cardiovasculaire est l’atteinte la plus grave chez les patients atteints du syndrome de Marfan.
La quantité insuffisante de fibrilline produite rend la paroi de l’aorte extrêmement fragile.
A savoir ! L’aorte est l’artère qui reçoit le sang du cœur et le redistribue dans tout l’organisme. Comme le cœur éjecte le sang par à-coups, l’aorte à tendance à se dilater avec le temps.
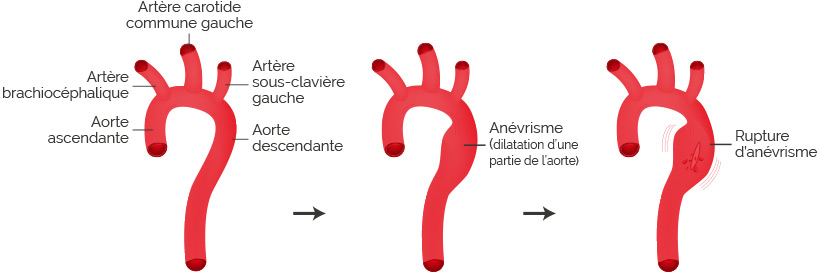 Compte tenu de cette fragilité de la paroi aortique, la dilatation aortique chez les patients Marfan devient plus rapide que chez les individus non touchés par le syndrome. Cette dilatation plus rapide peut aboutir à des complications graves telles qu’une insuffisance valvulaire aortique (fermeture incomplète des valvules aortiques, situées entre le ventricule gauche et l’aorte), un anévrysme aortique (dilatation d’une partie de l’aorte), voire même une dissection aortique (déchirure partielle de la paroi de l’aorte).
Compte tenu de cette fragilité de la paroi aortique, la dilatation aortique chez les patients Marfan devient plus rapide que chez les individus non touchés par le syndrome. Cette dilatation plus rapide peut aboutir à des complications graves telles qu’une insuffisance valvulaire aortique (fermeture incomplète des valvules aortiques, situées entre le ventricule gauche et l’aorte), un anévrysme aortique (dilatation d’une partie de l’aorte), voire même une dissection aortique (déchirure partielle de la paroi de l’aorte).
Quel que soit la complication, une intervention chirurgicale peut s’avérer nécessaire pour limiter les dégâts encourus et parfois le décès de l’individu.
Atteinte ophtalmologique
 L’œil est l’autre organe qui peut être atteint régulièrement par la maladie.
L’œil est l’autre organe qui peut être atteint régulièrement par la maladie.
Des complications oculaires, de degrés variables peuvent survenir mais parfois passer inaperçues, notamment chez les enfants :
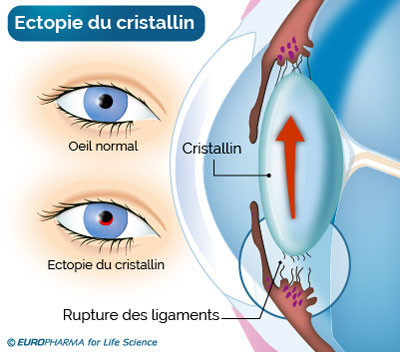
- Les déplacements du cristallin (ectopie, luxation) sont les symptômes oculaires les plus fréquemment rencontrés chez les malades de Marfan.
A savoir ! Le cristallin est la lentille qui se situe derrière la partie colorée de l’œil et qui permet la mise au point de la vision à des distances différentes. Il peut se déplacer (ectopie) ou se détacher (luxation). Cela peut se produire sur les deux yeux. Ce mouvement anormal du cristallin précède la myopie, l’œil n’étant plus capable d’apprécier les distances et donc de faire des mises au point.
- Un décollement de la rétine (couche qui tapisse le fond de l’œil et qui permet la formation des images) peut aussi se produire et donner naissance à une vision marquée par un voile noir ou gris.
- Enfin, la cataracte ou le glaucome semblent se développer plus rapidement et plus précocement chez les individus Marfan par rapport aux personnes non atteintes.
A savoir ! La cataracte est une opacification du cristallin qui perturbe progressivement la vision. Le glaucome correspond à une augmentation de la pression à l’intérieur de l’œil.
Atteinte squelettique
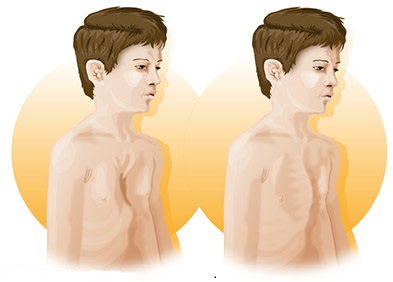
Les personnes atteintes du syndrome de Marfan sont souvent « anormalement » grandes et fines. C’est le symptôme physique le plus caractéristique de la maladie. Mais ces patients peuvent également souffrir de déformations plus classiques de la colonne vertébrale, comme des scolioses.
Les atteintes squelettiques majoritairement retrouvées chez les individus Marfan sont les suivants :
- Grande taille ;
- Croissance excessive des membres longs ;
- Maigreur ;
- Doigts longs et fins ;
- Déformations du tronc ;
- Déformations de la colonne vertébrales ;
- Déformation possible du sternum ;
- Hyperlaxité articulaire (élasticité très importante des tissus) conduisant souvent à des entorses ou à des douleurs articulaires touchant les chevilles et les pieds ;
- Modifications de la forme du visage : visage allongé, menton prononcé, palais étroit et « en ogive » (palais étroit en forme de V et non en forme U (forme normale)), chevauchement des dents.
Atteinte neurologique (ectasie durale)
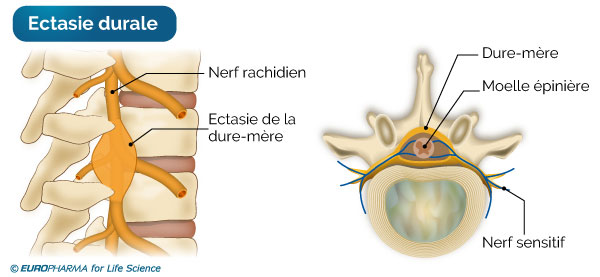 L’ectasie (dilatation) durale est la dilatation du sac (dural) qui contient la moelle épinière. Ce sac, qui est dans la colonne vertébrale, s’élargit vers le bas (niveau lombaire et sacré), là où la pression du liquide céphalo-rachidien est importante, dans le syndrome de Marfan, alors qu’il s’affine normalement chez le sujet normal.
L’ectasie (dilatation) durale est la dilatation du sac (dural) qui contient la moelle épinière. Ce sac, qui est dans la colonne vertébrale, s’élargit vers le bas (niveau lombaire et sacré), là où la pression du liquide céphalo-rachidien est importante, dans le syndrome de Marfan, alors qu’il s’affine normalement chez le sujet normal.
Cette atteinte d’ordre neurologique n’entraîne en aucun cas des symptômes physiques chez l’individu et peut être observée à l’IRM (Imagerie par Résonance Magnétique) et au scanner.
Atteinte pulmonaire
Les personnes atteintes par le syndrome de Marfan ont plus de risque de voir s’installer dans leurs poumons des bulles d’air, caractérisant ce que l’on appelle aussi l’emphysème pulmonaire.
Par ailleurs, lorsque ces bulles d’air sont rompues entre la plèvre (espace séparant le poumon de la paroi) et le poumon, cette rupture peut déclencher un pneumothorax (présence d’air dans la cage thoracique, ce qui compriment les poumons).
Le pneumothorax se présente par une douleur soudaine dans la poitrine et l’impression de ne pas pouvoir respirer. Les malades de Marfan ont un risque 50 fois plus élevé de développer un pneumothorax, comparé aux individus non atteints.
A savoir ! L’emphysème pulmonaire est une maladie pulmonaire se caractérisant par une dilatation des alvéoles pulmonaires.
Atteinte dermatologique
La peau est très fragilisée chez les individus Marfan, marquée par une plus grande finesse et le développement de vergetures apparaissant dès l’enfance (épaules, bas du dos).
Diagnostic du syndrome de Marfan
L’établissement du diagnostic est souvent difficile à poser étant donné la complexité des systèmes atteints par le syndrome de Marfan. Un ensemble de critères ont toutefois été établis pour aider à poser ce diagnostic. Ces critères sont essentiellement cliniques, basés sur l’atteinte des différents organes qui sont impliqués dans ce type de syndrome.
Il existe différentes tables de critères dont parmi elles, figurent les critères de Ghent (critères adaptés de Gand, 1996), présentés par exemple dans le tableau ci-dessous.
| Critères diagnostics du syndrome de Marfan selon Ghent | |||
| Système | Signes cliniques majeurs | Signes cliniques mineurs | Définition de l’atteinte du système |
| Squelettique | Pectus carinatum ou exclavatum, nécessitant une chirurgie
Rapport segment supérieur sur segment inférieur bas ou envergure sur taille > 1,05 Signe du poignet ou du pouce Scoliose > 20 ou spondylolisthésis Extension maximale des coudes < 170 Pied plat Protrusion acétabulaire |
Pectus excavatum modéré
Hyperlaxité ligamentaire Palais ogival avec chevauchement des dents Faciès caractéristique |
Majeure si au moins 4 signes cliniques majeurs sont présents |
| Oculaire | Ectopie cristallin | Cornée plate
Globe oculaire allongé Iris hypoplasique ou hypoplasie du muscle ciliaire |
Présence d’au moins deux signes mineurs |
| Cardio- vasculaire | – Dilatation de l’aorte ascen- dante intéressant les sinus de Valsalva
– Dissection aortique |
Insuffisance aortique
Prolapsus valvulaire mitral avec ou sans fuite Dilatation de l’artère pulmonaire avant l’âge de 40 ans Calcification de l’anneau mitral avant l’âge de 40 ans Anévrysme ou dissection de l’aorte abdominale avant l’âge de 50 ans |
Présence d’au moins d’un signe mineur |
| Pulmonaire | Pneumothorax spontané
Bulle apicale |
Présence d’au moins un signe mineur | |
| Cutané | Vergetures (à l’exclusion d’une grossesse, perte de poids)
Hernies récidivantes |
Présence d’au moins un signe mineur | |
| Dure-mère | Ectasie de la dure-mère lombo-sacrée | Présence du signe majeur | |
| Génétique | Un parent direct ayant les critères diagnostics
Mutation de FBN1 déjà connue pour provoquer un MFS |
||
Traitement et autres options thérapeutiques du syndrome de Marfan
A ce jour, il n’existe aucun traitement curatif du syndrome de Marfan, ni aucune façon de corriger l’altération du tissu conjonctif.
Aujourd’hui, l’objectif du traitement est de prévenir et de stabiliser les troubles avant l’apparition de complications potentiellement graves. Les bêtabloquants (comme l’aténolol et le propranolol) sont administrés pour faciliter le flux sanguin dans l’aorte. En présence d’une aorte dilatée ou d’un anévrisme, la partie altérée peut être réparée ou remplacée lors d’une intervention chirurgicale.
Par ailleurs, les femmes enceintes courent un risque plus important de complication au niveau de leur aorte, c’est pourquoi une réparation de l’aorte avant la conception doit être envisagée.
Le décollement du cristallin ou de la rétine peuvent, en général, être traités chirurgicalement.
Enfin, des séances de kinésithérapie peuvent aider à soulager d’éventuelles douleurs articulaires et peuvent nécessiter le port d’un corset lorsque les déformations de la colonne vertébrale ou du thorax sont déjà produites.
Lucie B., Biologiste spécialisée en E-santé
Cet article vous a-t-il été utile ?