Epidermolyse bulleuse
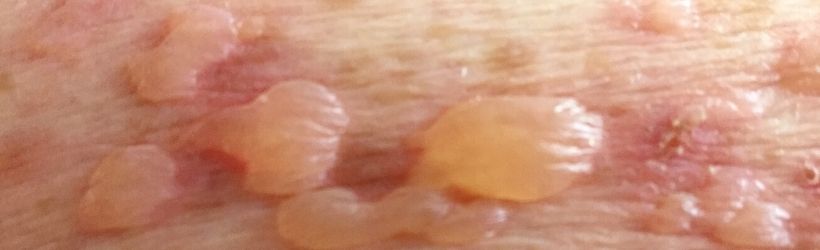
Les épidermolyses bulleuses touchent 1 nouveau-né sur 20 000, soit environ 30 000 cas en Europe et 500 000 dans le monde. Il existe plusieurs types d’épidermolyse bulleuse.
L’épidermolyse bulleuse acquise
Qu’est-ce que l’épidermolyse bulleuse acquise ?
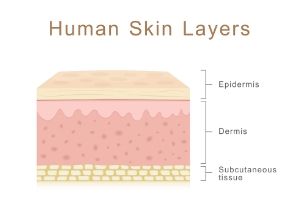
Une épidermolyse bulleuse acquise est une dermatose rare d’origine auto-immune. La maladie est due à la présence d’anticorps dirigés contre le collagène, constituant de la jonction dermo-épidermique. En effet, le dépôt des anticorps sur le collagène engendre un clivage entre l’épiderme et le derme, comme pour l’épidermolyse héréditaire qui elle est cependant due à une anomalie génétique.
Quels symptômes ?
La maladie peut se présenter sous deux formes : classique ou inflammatoire.
La forme classique se développe à l’âge adulte. Les bulles sont localisées sur la peau saine, elles sont flasques, tendues ou hémorragiques. Ces lésions sont provoquées par des traumatismes minimes, et prédominent sur les zones de frottement. Les muqueuses et les phanères sont également fréquemment atteints.
La forme inflammatoire ressemble à une autre pathologie : la pemphigoïde bulleuse. Chez l’enfant, elle se traduit par la présence de bulles sur une peau érythémateuse (rouge), des plaques d’urticaire (sans bulle) et des lésions diffuses qui ne se limitent pas aux zones de frottement. Dans 10 à 50% des cas, une pathologie est associée, par exemple la maladie de Crohn, la rectocolite hémorragique ou un diabète.
Quel diagnostic ?
Le diagnostic est évoqué devant les symptômes du patient. La confirmation du diagnostic repose sur l’étude des tissus via une biopsie.
L’évolution de l’épidermolyse bulleuse acquise est chronique, et la guérison très lente avec des cicatrices. Les formes inflammatoires peuvent évoluer en forme classique, tout comme les formes classiques peuvent présenter des poussées inflammatoires.
La gravité de la maladie dépend de la localisation des lésions, notamment oculaires et ORL qui peuvent engager le pronostic fonctionnel, voire vital.
Quel traitement ?
Le traitement de l’épidermolyse bulleuse acquise repose sur des soins de la peau et la prescription de certaines molécules, la dapsone et les corticoïdes. Chez les enfants, l’association des deux a montré des bénéfices. Chez l’adulte, des immunosuppresseurs (azathioprine, cyclosporine) sont également efficaces.
Les soins consistent à effectuer un nettoyage doux de la peau, prévenir les traumatismes cutanés, détecter et traiter les infections cutanées.
L’épidermolyse bulleuse héréditaire
Qu’est-ce que l’épidermolyse bulleuse héréditaire ?

L’épidermolyse bulleuse héréditaire regroupe en fait plusieurs pathologies génétiques rares caractérisées par une fragilité de la peau et des muqueuses. Cette fragilité se manifeste par la survenue de bulles ou d’érosions cutanées à l’occasion de traumatismes minimes de la peau ou survenant spontanément. Ces lésions sont assimilables à des brûlures, plus ou moins profondes, et douloureuses.
La maladie peut aussi bien se manifester dès la naissance qu’à la fin de l’enfance ou de l’adolescence. La gravité de l’épidermolyse est définie selon la fréquence des répétitions des lésions, la fragilité cutanée face aux traumatismes et la profondeur des lésions. Ces éléments dépendent de la forme génétique de la maladie. Ainsi, les différents types d’épidermolyses bulleuses héréditaires peuvent avoir une évolution et un pronostic très différent, allant d’une forme mineure à une forme létale.
L’épidermolyse bulleuse héréditaire est causée par des anomalies génétiques qui affectent des gènes codant diverses protéines impliquées dans l’intégrité structurelle et fonctionnelle des tissus cutanés. L’atteinte d’un même gène peut cependant aboutir à plusieurs types d’épidermolyse bulleuse héréditaire, avec des symptômes plus ou moins sévères. On distingue 3 grands types d’épidermolyse bulleuse héréditaire :
- Epidermolyse bulleuse simple, la plus fréquente (75 à 85% des cas). L’anomalie génétique affecte les gènes codant la kératine 5 ou 14. Le défaut de cohésion se situe entre les cellules de l’épiderme (couche cutanée superficielle) ;
- Epidermolyse bulleuse jonctionnelle lorsque l’anomalie est localisée à la jonction épiderme-derme (ou sous l’épiderme) ;
- Epidermolyse bulleuse dystrophique où l’anomalie est située au niveau du derme.
Ces maladies ont deux modes possibles de transmission :
- « dominante », la maladie s’exprime lorsqu’une seule des deux copies du gène est mutée. Ce mode de transmission suppose que l’un des deux parents est lui-même atteint ;
- « récessive », la maladie s’exprime lorsque les deux copies du gène sont anormales, chacune transmise par les deux parents.
Quels symptômes ?
L’épidermolyse bulleuse héréditaire se manifeste par la présence de vésicules, d’ulcérations, de croûtes et de cicatrices. Certaines formes de la maladie associent d’autres symptômes comme :
- Une atteinte des ongles ;
- Une alopécie ;
- Une hyperpigmentation ;
- Une kératodermie palmo-plantaire (épaississement de la peau) ;
- Une dystrophie musculaire ;
- Etc.
A noter ! Dans les formes sévères et généralisées, des complications peuvent survenir : malnutrition, douleur, contractures articulaires, inflammation chronique, carcinome cutané.
Les symptômes peuvent varier selon le type d’épidermolyse bulleuse héréditaire.
La forme simple qui est la plus répandue. Elle se manifeste par la présence de bulles au niveau des mains et des pieds dès la naissance ou dans les mois qui suivent. Ces bulles apparaissent suite à un choc, un frottement ou avec la chaleur. Les symptômes s’apaisent dans l’enfance ou l’adolescence, cependant une fragilité cutanée persiste.
La forme jonctionnelle regroupe un ensemble de pathologies assez variées. Certaines sont rapidement mortelles en raison des complications infectieuses et respiratoires, tandis que d’autres se rapprochent de la forme simple.
La forme dystrophique peut être :
- Elle est caractérisée par une éruption bulleuse généralisée (sauf au niveau des muqueuses). Les lésions sont plus fréquentes au niveau dans les zones de traumatismes. L’évolution est favorable avec le temps, cependant les bulles peuvent laisser des cicatrices.
- Récessive. Elle est responsable d’un handicap majeur. Les bulles se manifestent dès le moindre traumatisme, voire spontanément. Les atteintes muqueuses sont fréquentes, et peuvent engendrer des difficultés pour s’alimenter. Les lésions peuvent entraîner une fusion des doigts et des orteils, et une rétractation cutanée pouvant engendrer un handicap fonctionnel important. Une chute des cheveux est souvent associée, avec un retentissement psychologique. L’importance des plaies peut engendrer deux grands types de complications : une surinfection bactérienne et un cancer cutané. Enfin, une ostéoporose sévère est généralement présente, et à l’origine d’importantes douleurs.
Quel diagnostic ?
Le diagnostic est évoqué devant les symptômes du patient. La confirmation du diagnostic repose sur l’étude des tissus via une biopsie.
Quel traitement ?
Aucun traitement ne permet de traiter la cause génétique de cette pathologie. La prise en charge est symptomatique.
Les soins infirmiers quotidiens consistent à percer les nouvelles bulles avec une désinfection des plaies, à réaliser un bain quotidien et à appliquer des pansements gras non collants.
L’antisepsie doit être rigoureuse afin de limiter le risque de surinfection des lésions. Une pommade antibiotique doit être appliquée en cas de signe d’infection débutante.
La kinésithérapie permet de limiter les éventuelles difficultés à la marche (en lien avec un épaississement cutané au niveau de la plante des pieds). Un traitement orthopédique sert à éviter certaines rétractations ou à corriger la fusion des doigts.
Pour la douleur, des antalgiques plus ou moins puissants, selon l’importance des symptômes sont prescrits.
Charline D., Docteur en pharmacie
– Epidermolyse bulleuse acquise. LE MANUEL MSD. Consulté le 6 février 2020.
– Epidermolysis bullosa acquisita. ORPHANET. Consulté le 6 février 2020.
Cet article vous a-t-il été utile ?