Maladie de Fabry
La maladie de Fabry est une pathologie héréditaire rare causée par une anomalie génétique. Ainsi, elle concerne toute la famille puisqu’elle se transmet de génération en génération. Cette maladie est chronique et atteint progressivement divers organes.
Définition
La maladie de Fabry a été décrite pour la première fois en 1898 par deux médecins : l’un anglais, William Anderson et l’autre allemand, Johann Fabry. D’ailleurs, la maladie est parfois appelée « la maladie de Anderson-Fabry ».
Cette pathologie est une maladie héréditaire rare. Le nombre de nouveaux cas par an dans le monde est évalué à 1 naissance sur 80 000 et le nombre de patients atteints de la maladie à près d’1 individu sur 3000. Enfin, elle concerne tant les hommes que les femmes.
La maladie de Fabry est une pathologie dite de « surcharge lysosomale » résultant d’une déficience enzymatique. Une enzyme est une molécule indispensable au bon déroulement des réactions chimiques se produisant dans l’organisme. En fait, les patients souffrants de cette maladie ne possèdent pas ou produisent en quantité insuffisante l’a-galactosidase A qui est une enzyme importante pour la bonne élimination de certains déchets. Sans cette dernière, les déchets ne sont pas évacués de l’organisme et s’accumulent dans les cellules des principaux organes comme le rein, le cœur ou le système nerveux. Or, cette accumulation de matière perturbe le bon fonctionnement des organes.
À savoir ! L’α-galactosidase A est d’ordinaire présente dans les lysosomes. Les lysosomes sont de petits centres de recyclage présents dans nos cellules. Ils assurent la dégradation des déchets. Lorsque cette enzyme est absente, les déchets s’accumulent. Le principal d’entre eux s’appelle globotriaosylcéramide ou GL-3.
La maladie est héréditaire, ce qui sous-entend qu’elle est engendrée par une anomalie chromosomique. Le gène défectueux est plus particulièrement situé sur le chromosome X. En effet, nous héritons tous un chromosome X de notre mère et un chromosome X de notre père, auquel cas, on est une femme, ou un homme lorsque notre père nous transmet un chromosome Y. En effet, le sexe féminin est définit comme étant XX tandis que le sexe masculin est XY.
Or, le gène défectueux est situé sur le chromosome X, ce qui implique que tant les femmes que les hommes peuvent posséder ce gène (puisque les deux possèdent au minimum un X).
Un homme atteint de la maladie transmettra donc (puisque le seul X dont il dispose est défectueux) automatiquement le gène défectueux à ses filles, mais pas à ses fils.
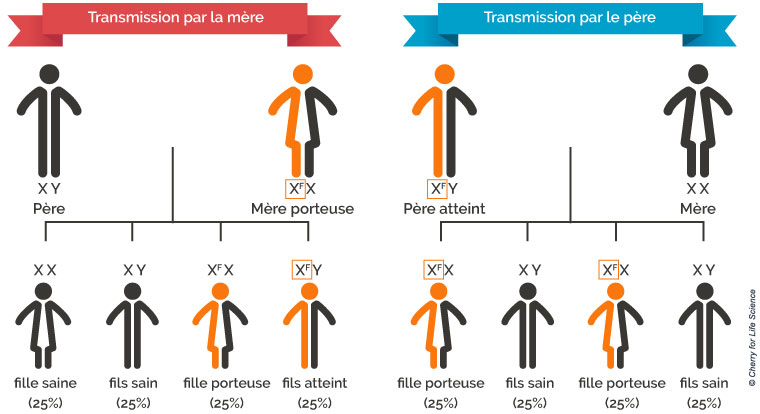
À savoir ! Les femmes atteintes de la maladie de Fabry sont souvent qualifiées de « porteuses ». Ce terme indiquerait normalement qu’elles ne présentent pas de symptôme. Or, c’est faux ! Tout comme les hommes possédant un chromosome défectueux, les femmes sont atteintes de la maladie. En revanche, leurs symptômes sont plus tardifs.
Symptômes de la maladie de Fabry
Le déchet GP-3 s’accumule dans les cellules du corps entier. Les symptômes de la maladie peuvent alors être très divers et s’aggravent généralement avec le temps. En effet, tous les patients atteints de la maladie de Fabry ne présentent pas les mêmes symptômes. Par ailleurs, la gravité de la pathologie varie d’une personne à une autre et les symptômes peuvent même différer selon l’évolution de la maladie.
Il est donc difficile de décrire une forme « typique ». En effet, le tableau clinique de cette pathologie peut recouvrir tout un spectre de sévérité allant des formes légères aux formes plus graves. En effet, la gravité et la vitesse d’apparition des symptômes de la maladie dépendent de la quantité d’enzyme présente dans l’organisme.
Les formes graves peuvent présenter toutes les manifestations caractéristiques telles que des atteintes : neurologiques (douleurs), dermatologiques (angiokératomes), rénales (insuffisance rénale), cardiovasculaire (arythmie, cardiomyopathie), cochléo-vestibulaires et cérébro-vasculaires (AVC).
La douleur est un symptôme fréquent au début de la maladie. Elle se manifeste par des paresthésies (troubles de la sensibilité pouvant se traduire par des fourmillements) et des sensations de brûlure à l’origine de crises très douloureuses. Cependant, elle peut disparaître une fois l’âge adulte atteint.
Une anhidrose ou hypohidrose (absence ou diminution de la transpiration) peut être présente. Elle engendre souvent une intolérance à la chaleur et à l’effort physique.
D’autres symptômes existent :
- Des modifications de la cornée ;
- Des acouphènes ;
- Une fatigue chronique ;
- Des anomalies cardiaques ;
- Des anomalies cérébro-vasculaires (arythmie, angine de poitrine par exemple) ;
- Une dyspnée (difficulté à respirer) ;
- Une atteinte des reins.
Avec l’âge, l’aggravation des symptômes peut conduire à une défaillance de plusieurs organes, par exemple une insuffisance rénale terminale ou des complications cérébro-vasculaires ou cardio-vasculaires mettant en jeu le pronostic vital. Cette pathologie réduit l’espérance de vie des patients de près de 20 ans pour les hommes et de 10 ans pour les femmes.
Diagnostic de la maladie de Fabry
Un diagnostic précoce est important afin de limiter l’évolution de la maladie et d’améliorer la qualité de vie des patients.
La majorité des patients atteints de la maladie de Fabry ressentent les premières manifestations dès le début de l’enfance. Chez l’enfant, les symptômes les plus fréquents sont les douleurs et la difficulté à supporter la chaleur lors d’un exercice physique.
Certaines pathologies (sclérose en plaques chez l’adulte, polyarthrite rhumatoïde ou douleurs de croissance chez l’enfant) peuvent provoquer les mêmes types de symptômes, il est alors nécessaire de les écarter au préalable.
Le diagnostic de la maladie de Fabry est effectué en laboratoire et repose sur la démonstration du déficit enzymatique dans le sang ou les urines. Chez les femmes, il est possible de faire une analyse ADN en complément.
À savoir ! Un diagnostic prénatal ou préimplantatoire est possible.
Traitement de la maladie de Fabry
Depuis le début des années 2000, un traitement spécifique de la maladie de Fabry a été mis au point : l’enzymothérapie substitutive. Des études prometteuses sont en cours. Cette thérapie repose sur l’utilisation d’une a-galactosidase A produite in vitro et introduite dans l’organisme afin de palier au manque enzymatique. Elle est administrée toutes les 2 semaines par perfusion. De manière générale, elle est très bien tolérée par les patients et présente peu d’effets secondaires. En effet, lors de la perfusion, le patient peut ressentir des frissons, une impression de variation de la température, de la fièvre, des nausées et des maux de tête. Par ailleurs, à ce stade des connaissances sur la pathologie et compte tenu de son caractère progressif, on peut penser que plus la thérapie est débutée rapidement, meilleurs seront les résultats. Il semblerait également que l’enzymothérapie ait plus de chance de succès lorsque les complications ne sont pas encore présentes et à condition qu’elle soit administrée régulièrement.
Finalement, traitée précocement et à dose efficace, cette thérapie peut ralentir voire stopper l’évolution de la maladie et éviter les complications offrant ainsi une meilleure qualité de vie aux patients.
La prise en charge de la maladie repose également sur un traitement antidouleur et la prévention des complications, particulièrement cardiaques (anti arythmiques, pacemakers ou défibrillateurs implantables) et rénales (médicaments, dialyse, greffe).
Charline D., Pharmacien
– Maladie de Fabry. Orphanet. Consulté le 27 mars 2018.
Cet article vous a-t-il été utile ?